

ในปัจจุบัน ปัญญาประดิษฐ์เข้ามามีบทบาทในการสร้าง generative model จำนวนมาก ไม่ว่าจะการสร้างรูป เสียง เพลง หรือคำบรรยายในวีดีโอ รวมถึงในเภสัชศาสตร์ เราก็สามารถใช้ปัญญาประดิษฐ์ในการช่วยออกแบบโมเลกุลได้ด้วยเช่นกัน โดยในบทความนี้ จะพูดถึงการใช้การเรียนรู้เชิงลึกหรือ deep learning ว่าจะสามารถเข้ามาช่วยในการออกแบบโมเลกุลเพื่อพัฒนาต่อเป็นยาอย่างไรได้บ้าง
โดยทั่วไปแล้ว โมเลกุลคือกลุ่มก้อนของอะตอมที่เชื่อมต่อกันด้วยพันธะ หรืออาจเขียนให้อยู่ในรูปสูตรโมเลกุลอย่างเช่น C6H6O12 แต่หลายคนอาจยังไม่ทราบว่า โมเลกุลมีการ representation อีกหลายรูปแบบ ทั้งรูปภาพสองมิติ แบบจำลองสามมิติ กราฟ ECFP (Extended Connectivity Fingerprints) และหนึ่งในนั้นคือ Simplified Molecular Input Line Entry System หรือเราสามารถเรียกสั้น ๆ ว่า SMILES โดยการ representation โมเลกุลแบบ SMILES นี้จะมีลักษณะเป็น line notation ช่วยให้เราสามารถมองโครงสร้างเคมีเป็น text ธรรมดาที่มีการใช้ตัวอักษรภาษาอังกฤษแทนอะตอมต่าง ๆ และใช้สัญลักษณ์แทนพันธะ เช่น – แทนพันธะเดี่ยว, = แทนพันธะคู่, # แทนพันธะสาม เป็นต้น ตัวอย่างเช่น เอทิลีน (C2H4) เมื่อแสดงในรูปแบบ SMILES จะเป็น C=C [1] หรือยา Ciprofloxacin เมื่อเขียนให้อยู่ในรูป SMILES จะได้ดังแสดงในรูปที่ 1 โดยฐานข้อมูลหลัก ๆ ที่มีการเก็บข้อมูลโมเลกุลเคมีและมักถูกนำไปเป็นข้อมูลในการสร้างโมเดล ได้แก่ ChEMBL, ZINC, Pubchem เป็นต้น
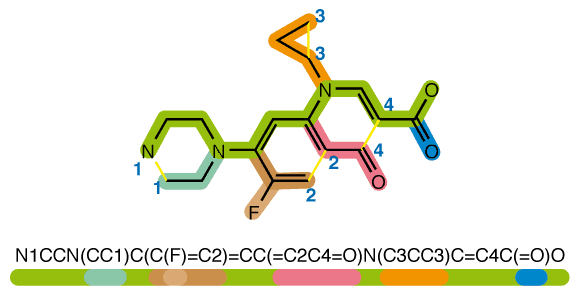
(https://en.wikipedia.org/wiki/Simplified_molecular-input_line-entry_system#/media/File:SMILES.png)
ในการเตรียมข้อมูลโครงสร้างทางเคมีในรูปแบบ SMILES มีขั้นตอนดังนี้
- ทำการสร้าง corpus หรือคลังข้อมูลของตัวอักขระทั้งหมดใน SMILES ที่ประกอบด้วย ตัวอักษร ตัวเลข และสัญลักษณ์ จากนั้น mapping ตัวอักขระให้เข้ากับตัวเลขเพื่อทำเป็น index ประจำอักขระแต่ละตัว รวมถึงเพิ่มอักขระที่แสดงถึงจุดสิ้นสุดของโมเลกุล (End of Sentence) โดยใช้ตัวอักขระที่ไม่มีใน SMILES เช่น “\n”
- เพิ่มจุดสุดท้ายของโมเลกุลยาแต่ละตัว เพื่อให้โมเดลเรียนรู้ว่าจะจบโมเลกุลที่ใด รวมถึงเป็นตัวกำหนดจุดสิ้นสุดของโมเลกุลในตอนที่ใช้โมเดลออกแบบโมเลกุลขึ้นใหม่
- ทำการเข้ารหัสแบบ one-hot (one-hot encoding) โดยการแจกแจงข้อมูลประเภทหมวดหมู่ให้อยู่ในรูปแบบคอลัมน์ และปรับข้อมูลข้อในให้เป็น Binary values หรือมีค่าเพียง 1 หรือ 0 เท่านั้น โดย 1 จะแสดงถึงการใช้ตัวอักขระนั้น และ 0 จะแสดงถึงการไม่ได้ใช้ ดังแสดงในรูปที่ 2 เพื่อให้อยู่ในรูปของเวกเตอร์และสามารถนำไปใช้ประมวลผลในโมเดลได้
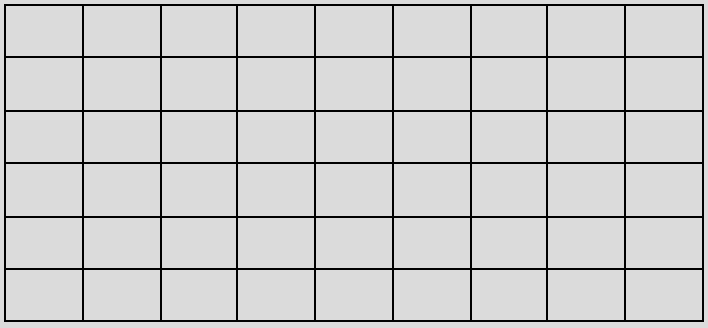
| C | 1 | 0 | 0 | 1 | 0 | 0 | 0 | … | 0 |
| N | 0 | 1 | 0 | 0 | 0 | 0 | 0 | … | 0 |
| = | 0 | 0 | 1 | 0 | 1 | 0 | 0 | … | 0 |
| O | 0 | 0 | 0 | 0 | 0 | 1 | 0 | … | 0 |
| … | … | … | … | … | … | … | … | … | … |
| \n | 0 | 0 | 0 | 0 | 0 | 0 | 1 | … | 1 |
| C | N | = | C | = | O | \n | … | \n |
ข้อมูลหลังจากปรับให้อยู่ในรูปเวกเตอร์แล้วจะมีทั้งหมด 3 มิติด้วยกัน คือ จำนวนอักขระในคลัง x ความยาวสูงสุดของอักขระในโมเลกุลในชุดข้อมูล x จำนวนโมเลกุลทั้งหมด ซึ่งความยาวสูงสุดของอักขระในโมเลกุลค่อนข้างมีผลกระทบสูงกับขนาดของข้อมูล ดังนั้น หากต้องการลดขนาดของข้อมูลลง วิธีหนึ่งที่สามารถทำได้และไม่กระทบกับข้อมูลภาพรวมมาก คือ การกำหนดความยาวสูงสุดของอักขระในโมเลกุล เนื่องจากในฐานข้อมูลมีโมเลกุลยาจำนวนมาก อย่างในฐานข้อมูล ChEMBL29 มีจำนวนถึง 2.1 ล้านโมเลกุล ซึ่งโมเลกุลบางตัวมีความยาว 200 ตัวอักษร แต่ความยาวเฉลี่ยของโมเลกุลจากทั้งฐานข้อมูลมีเพียง 60 ตัวอักษรเท่านั้น ในการกำหนดความยาวของอักขระ อาจใช้หลักทางสถิติมาช่วย เช่น กฎเชิงประจักษ์ (Empirical Rule) เพื่อตัด outliner ออก หรือกำหนดตามค่าที่ต้องการ ซึ่งการตัดโมเลกุลยาบางตัวที่มีความยาวมากเกินไปจะช่วยลดภาระของหน่วยความจำ และช่วยให้โปรแกรมประมวลผลได้เร็วขึ้นในตอนที่โมเดลทำการเรียนรู้
ในการสร้าง generative model สำหรับออกแบบโมเลกุลยา มีงานวิจัยจำนวนมากที่ได้นำการเรียนรู้เชิงลึกมาสร้าง ซึ่งโครงสร้างโมเดลสามารถสร้างได้หลากหลายรูปแบบ เช่น Recurrent Neural Network (RNN) [2][3], Autoencoder [4], Generative Adversarial Network (GAN) [5], Reinforcement Learning [6] เป็นต้น โดยในบทความนี้จะกล่าวถึงงานวิจัยของ Segler et al. [2] ที่นำ Long Short-Term Memory (LSTM) [7] ซึ่งเป็น RNN รูปแบบหนึ่งมาช่วยในการออกแบบโมเลกุลยา
ในการสร้างโมเดลสำหรับออกแบบโมเลกุล ข้อมูลประเภท SMILES มีสิ่งหนึ่งที่ต้องให้ความสำคัญคือ วงเล็บ โดยจะมีทั้งหมดสองประเภท คือ ( ) และ [ ] ซึ่งจำเป็นต้องใช้โครงสร้างโมเดลที่มีความสามารถเพียงพอในการเรียนรู้รูปแบบของวงเล็บว่ามีการเปิดปิดครบหรือไม่ เพราะถ้าไม่ครบหรือเกิน จะเป็นจุดหนึ่งที่ทำให้โมเลกุลที่ถูกออกแบบออกมามีโครงสร้างที่ไม่ถูกต้อง และไม่สามารถนำไปใช้งานต่อได้ ดังนั้น โมเดลที่ใช้จะต้องมีความสามารถในการติดตามการเปิดปิดของวงเล็บที่อาจอยู่ในตำแหน่งที่มีระยะห่างกันมาก โดยการงานวิจัยนี้ [2] ได้นำ LSTM ที่มีลักษณะการทำงานเหมือน RNN คือทำงานในรูปแบบวนซ้ำดังชื่อ โดยจะนำ output จาก state ก่อนหน้ามาเป็น input ใน state ควบคู่กับ input ณ state นั้น ๆ แต่มีเพิ่มในส่วนของ gate ที่ทำหน้าที่ควบคุมการไหลของข้อมูล ช่วยให้โมเดลทราบว่า ข้อมูลใดที่ควรเก็บหรือควรลบ และ cell state ที่เป็นตัวเก็บสถานะของ memory cell ทำให้ LSTM มีความสามารถในจดจำข้อมูลได้ในระยะยาวกว่า RNN รวมถึงสามารถติดตามการเปิดปิดของวงเล็บได้ ซึ่งเมื่อสร้างโมเดลเรียบร้อย ก็ทำการเทรนด้วยข้อมูลโมเลกุลที่ได้ทำการเตรียมข้อมูลไว้แล้วในข้างต้น
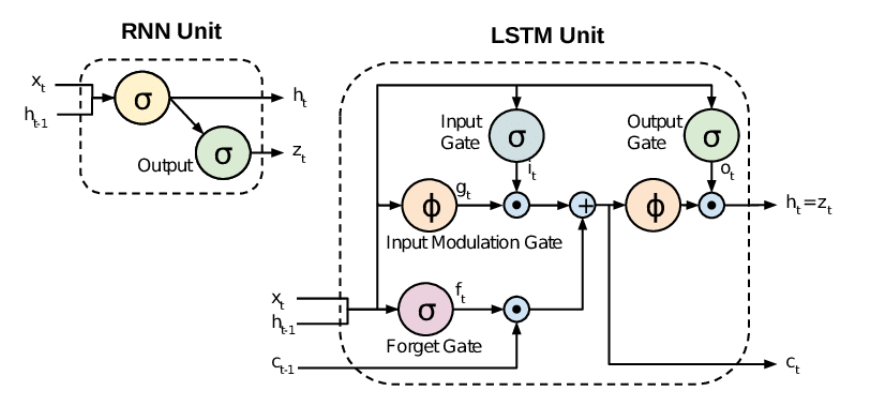
(https://ashutoshtripathicom.files.wordpress.com/2021/06/rnn-vs-lstm.png)
การใช้โมเดลในการออกแบบโมเลกุลยา จากรูปที่ 3 สมมติให้มีอักขระในคลังทั้งหมด 3 ตัว ได้แก่ c, 1 และ \n เริ่มจากการสุ่มอักขระเริ่มต้น s1 ซึ่งได้ออกมาเป็น c จากนั้น มีการปรับให้อยู่ในรูปของเวกเตอร์ x1 โดยใช้การเข้ารหัสแบบ one-hot ซึ่งจะได้เป็น (1, 0, 0) โดย 1 ตัวแรกแสดงถึง c และ 0 สองตัวหลังแสดงถึง 1 และ \n ตามลำดับ จากนั้นจึงนำเวกเตอร์นั้นไปเข้าโมเดล โมเดลจะทำการอัพเดทสถานะภายในตัวเอง (h1) และสร้าง output ออกมาเป็น y1 หรือเป็นการแจกแจงความน่าจะเป็น (Probability Distribution) ของอักขระถัดไป ซึ่งอักขระแรกที่ถูกสร้างใน t = 1 คือ s2 = 1 จากนั้น s2 จะถูกนำไปเข้ารหัสให้อยู่ในรูป x2 และนำเข้าโมเดลเพื่อสร้างอักขระตัวถัดไป โดยโมเดลจะทำการสร้างตัวถัดไปเรื่อย ๆ จนกว่าจะเจอ ‘\n’ หรือจุดสิ้นสุดของ SMILES และได้ c1ccccc1\n ออกมา โดยสถานะ h ที่อยู่ภายในโมเดลจะช่วยในการติดตามวงเล็บของอักขระที่ถูกสร้างออกมา [2]

(Segler et al., 2017)
หนึ่งในประโยชน์ของการออกแบบโมเลกุลคือในสายงาน drug discovery โดยปกติแล้วอาศัยการออกแบบโมเลกุลให้เฉพาะเจาะจงกับเป้าหมายที่ต้องการ ซึ่งทำได้ยากเนื่องจากโมเลกุลประจำแต่ละเป้าหมายนั้นมีข้อมูลน้อย ไม่เพียงพอให้โมเดลจดจำรูปแบบและทำการสร้างได้อย่างถูกต้อง ดังนั้น งานวิจัยส่วนมากจึงนิยมใช้การถ่ายทอดการเรียนรู้ (transfer learning) คือทำการสอนโมเดลด้วยโมเลกุลทั้งหมดในฐานข้อมูลก่อน จากนั้นค่อยทำการสอนโมเดลอีกครั้งแต่เลือกเฉพาะข้อมูลโมเลกุลที่ออกฤทธิ์กับเป้าหมายที่ต้องการ ซึ่งช่วยให้โมเดลสามารถออกแบบโมเลกุลที่สามารถออกฤทธิ์เจาะจงกับเป้าหมายได้ [2]
หลังจากได้โมเลกุลที่ออกแบบมาแล้ว จะต้องมีการตรวจสอบความถูกต้องของผลลัพธ์เหมือนโมเดลอื่น ๆ โดยเกณฑ์หลัก ๆ ที่จำเป็น ได้แก่
- ความถูกต้อง (Validity): โมเลกุลที่ออกแบบมีโครงสร้างถูกต้องตามหลักทางเคมี รวมถึงคุณสมบัติเหมือนตัวยา (Drug-likeness Property) เช่น คุณสมบัติตามกฎของ Lipinski (Lipinski’s Rule of Five) [8] ได้แก่ น้ำหนักโมเลกุลควรน้อยกว่าหรือเท่ากับ 500, ค่า CLogP ควรน้อยกว่าหรือเท่ากับ 5, จำนวนตัวให้พันธะไฮโดรเจนน้อยกว่าหรือเท่ากับ 5 และจำนวนตัวรับพันธะไฮโดรเจนน้อยกว่าหรือเท่ากับ 10 ซึ่งคุณสมบัติเหล่านี้สามารถตรวจสอบได้โดยใช้ RDKit [9] ซึ่งเป็น library ที่ใช้ประมวลผลเกี่ยวกับข้อมูลทางโมเลกุลยา
- ความเฉพาะตัว (Uniqueness): โมเดลสามารถออกแบบโมเลกุลได้ไม่ซ้ำกัน
- ความแปลกใหม่ (Novelty): โมเลกุลที่ออกแบบมีความใหม่ และไม่ซ้ำกับชุดข้อมูลที่นำมาเทรน
References
[1] Weininger, D. (1988). SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. Journal of Chemical Information and Modeling, 28(1), 31–36.doi:10.1021/ci00057a005
[2] Segler, M. H. S., Kogej, T., Tyrchan, C., & Waller, M. P. (2017). Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Central Science, 4(1), 120–131.doi:10.1021/acscentsci.7b00512
[3] Grisoni, F., Moret, M., Lingwood, R., & Schneider, G. (2020). Bidirectional Molecule Generation with Recurrent Neural Networks. Journal of Chemical Information and Modeling.doi:10.1021/acs.jcim.9b00943
[4] Blaschke, T., Olivecrona, M., Engkvist, O., Bajorath, J., & Chen, H. (2017). Application of Generative Autoencoder in De Novo Molecular Design. Molecular Informatics, 37(1-2), 1700123.doi:10.1002/minf.201700123
[5] De Cao, N., & Kipf, T. (2018). MolGAN: An implicit generative model for small molecular graphs. ArXiv, abs/1805.11973.
[6] Popova, M., Isayev, O., & Tropsha, A. (2018). Deep reinforcement learning for de novo drug design. Science Advances, 4(7), eaap7885.doi:10.1126/sciadv.aap7885
[7] Hochreiter, S.; Schmidhuber, J. (1997). Long short-term memory Neural computation. 9, 1735– 1780 DOI: 10.1162/neco.1997.9.8.1735
[8] Lipinski, C. A., Lombardo, F., Dominy, B. W. and Feeney, P. J. (2001). Experimental and computational approach to estimate solubility and permeability in drug discovery and development settings, Advanced Drug Delivery Reviews, 46, 3-26.
[9] RDKit: Open-source cheminformatics; http://www.rdkit.org
By Nutaya Pravalphruekul (Pear)


{kind=link}
{kind=link}